
Scientists developing innovative pharmaceuticals face the complex challenge of identifying molecules that can effectively bind to disease-causing proteins and modify their behavior. Understanding the precise three-dimensional configuration of these molecules is essential for predicting their interaction with specific protein surfaces.
However, a single molecule can fold into thousands of potential configurations, making experimental determination of these structures an incredibly time-consuming and costly endeavor—akin to finding a needle in a molecular haystack.
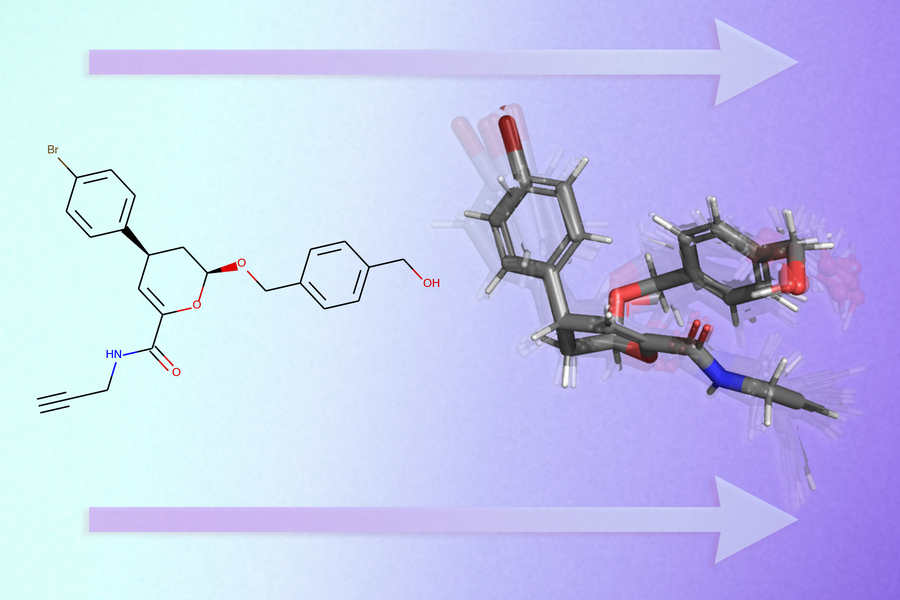
Researchers at MIT are harnessing the power of artificial intelligence to transform this intricate process. Their team has developed an advanced deep learning model that accurately forecasts the 3D conformations of molecules using only their 2D molecular structure graphs. This AI molecular structure prediction approach represents a significant leap forward in computational chemistry.
Their innovative system, GeoMol, processes molecular structures in mere seconds and outperforms existing machine learning models, including several commercial alternatives. This deep learning drug discovery tool could enable pharmaceutical companies to dramatically accelerate their research pipeline by substantially reducing the number of molecules requiring laboratory testing, explains Octavian-Eugen Ganea, a postdoc in the Computer Science and Artificial Intelligence Laboratory (CSAIL) and co-lead author of the paper.
"When analyzing how these molecular structures move in three-dimensional space, only specific components—the rotatable bonds—exhibit flexibility. Our key innovation involves modeling conformational flexibility from a chemical engineering perspective. Essentially, we're predicting the potential distribution of these rotatable bonds throughout the structure," notes Lagnajit Pattanaik, a graduate student in the Department of Chemical Engineering and co-lead author of the paper.
The research team includes Connor W. Coley, the Henri Slezynger Career Development Assistant Professor of Chemical Engineering; Regina Barzilay, the School of Engineering Distinguished Professor for AI and Health in CSAIL; Klavs F. Jensen, the Warren K. Lewis Professor of Chemical Engineering; William H. Green, the Hoyt C. Hottel Professor in Chemical Engineering; and senior author Tommi S. Jaakkola, the Thomas Siebel Professor of Electrical Engineering in CSAIL and a member of the Institute for Data, Systems, and Society. The findings were presented at the Conference on Neural Information Processing Systems.
Decoding Molecular Architecture
In molecular graphs, atoms are represented as nodes while the chemical bonds connecting them form edges. GeoMol utilizes a cutting-edge deep learning technique known as message passing neural networks, specifically designed for graph-based data processing. The researchers adapted this approach to predict critical aspects of molecular geometry with remarkable accuracy.
When presented with a molecular graph, GeoMol first predicts the lengths of chemical bonds between atoms and the angles formed by these bonds. The atomic arrangement and connectivity determine which bonds possess rotational capability.
The machine learning 3D molecular modeling system then predicts each atom's local structural environment individually, assembling neighboring rotatable bond pairs by calculating torsion angles and aligning them accordingly. A torsion angle defines the movement of three connected segments—in this case, three chemical bonds linking four atoms.
"The rotatable bonds can assume an extensive range of possible values. Our message passing neural network approach captures both local and global environmental factors influencing these predictions. Since rotatable bonds can adopt multiple configurations, we designed our prediction methodology to reflect this underlying distribution," Pattanaik explains.
Addressing Molecular Complexity
One significant challenge in predicting molecular 3D structures involves modeling chirality—the property where a molecule cannot be superimposed on its mirror image, similar to human hands. Chiral molecules interact differently with their environment, which can cause medications to bind incorrectly to proteins, potentially leading to harmful side effects.
Current machine learning methods typically require extensive optimization processes to properly identify chirality. However, GeoMol determines the 3D structure of each bond individually, explicitly defining chirality during prediction and eliminating the need for subsequent optimization.
After completing these predictions, GeoMol generates a set of probable 3D molecular conformations.
"We can now integrate our model end-to-end with systems that predict molecular attachment to specific protein surfaces. Unlike separate pipeline approaches, our GeoMol AI pharmaceutical research tool seamlessly integrates with other deep learning models," Ganea states.
Exceptional Performance Metrics
The researchers evaluated their model using a molecular dataset developed by Rafael Gomez-Bombarelli, the Jeffrey Cheah Career Development Chair in Engineering, and graduate student Simon Axelrod, which included molecules and their potential 3D conformations.
They assessed how many of these probable 3D structures their model could capture compared to other machine learning approaches and traditional methods.
Across nearly all evaluation metrics, GeoMol consistently outperformed competing models.
"We were thrilled to discover that our model operates at exceptional speeds. Importantly, while most algorithms slow significantly as rotatable bonds increase, we observed minimal performance degradation. This scalability with respect to rotatable bond count is promising for future applications, particularly for rapid prediction of 3D structures within proteins," Pattanaik remarks.
Looking ahead, the research team aims to apply GeoMol to high-throughput virtual screening, using the model to identify small molecule structures that would interact with specific proteins. They also plan to enhance GeoMol's capabilities through additional training data to improve prediction accuracy for larger molecules with numerous flexible bonds.
"Conformational analysis is fundamental to numerous computer-aided drug design tasks and represents a critical component in advancing machine learning approaches in pharmaceutical discovery," says Pat Walters, senior vice president of computation at Relay Therapeutics, who was not involved in this research. "I'm excited by the continuing innovations in this field and commend MIT for contributing to these broader scientific advancements."
This research was funded by the Machine Learning for Pharmaceutical Discovery and Synthesis consortium.

